Research
I believe that a fundamental understanding of the rules that govern natural biological processes is essential to rationally designer better and new technologies. With advanced computational tools, we have access to macroscopic time and length and data scales that allows us to better understand the complex building blocks of nature. Using machine learning, statistical mechanics, physics, and biology, we can link microscopic phenomena to macroscopic behavior.
My work has leverages these tools to understand and uncover principle guiding rules in biological design.
Computational Tools
Deep Learning

Generative Design
Deep learning is a subclass of machine learning approaches which utilize neural networks to learn complex patterns and relationships in large datasets. The term "deep" typically refers to the number of hidden layers in a neural network.
Generative models are a type of model which use learned patterns and characteristics to generate instances of new data of similar to that of the original dataset.
Alamdari et al., protein sequence and representative structure generated from deep generative model
Molecular Dynamics

Enhanced Sampling Simulations
Molecular dynamics is a powerful computational method that allows us to simulate the dynamics of biological molecules. In classical molecular dynamics a simple physics-based approach is used to describe the potential energy of a system. For example, the energy of a bond can be described by a simple harmonic potential, where the spring constant can be used to tune the interactions between different atom types. This simple idea can be expanded to study entire proteins, which are some of the most complex microscopic systems that exist in nature
Often times the phenomena we are interested in exploring occurs on timescales much longer than accessible by computers (> 1 microsecond). In these cases we can apply enhanced sampling methods, which are advanced computational tools, that allow us to explore underlying thermodynamic and kinetic driving forces in our systems.
Alamdari, bond vibration gif
Protein Design
Protein generation with evolutionary diffusion: sequence is all you need
Sarah Alamdari, Nitya Thakkar, Rianne van den Berg, Alex X. Lu, Nicolo Fusi, Ava P. Amini, Kevin K. Yang
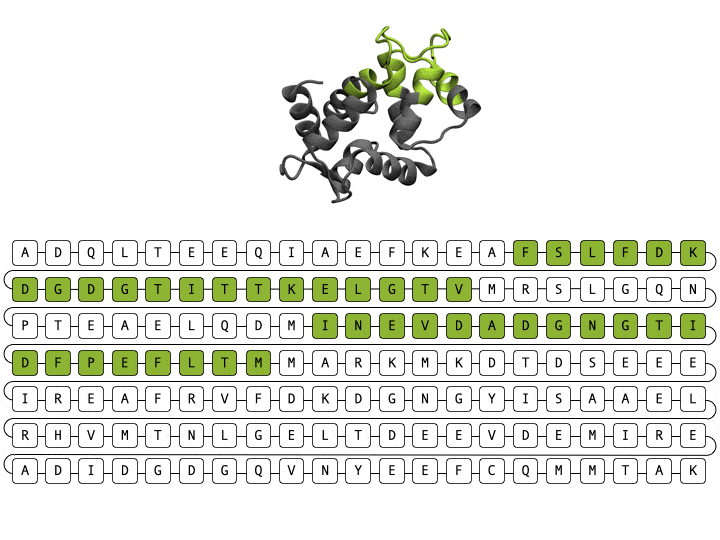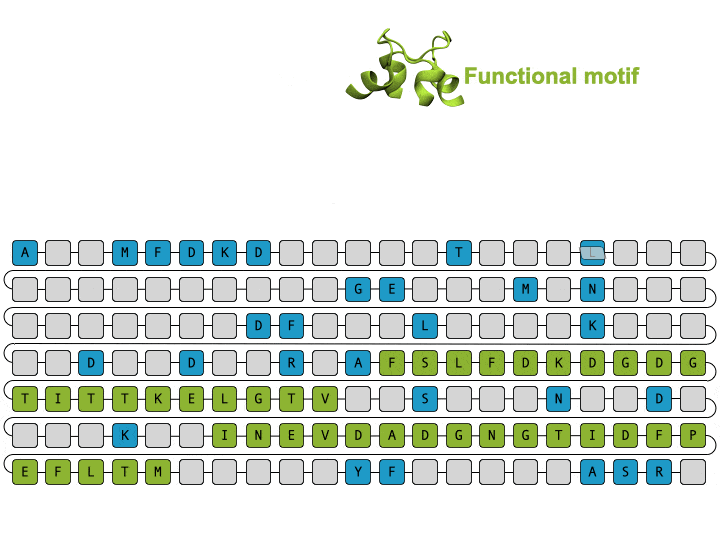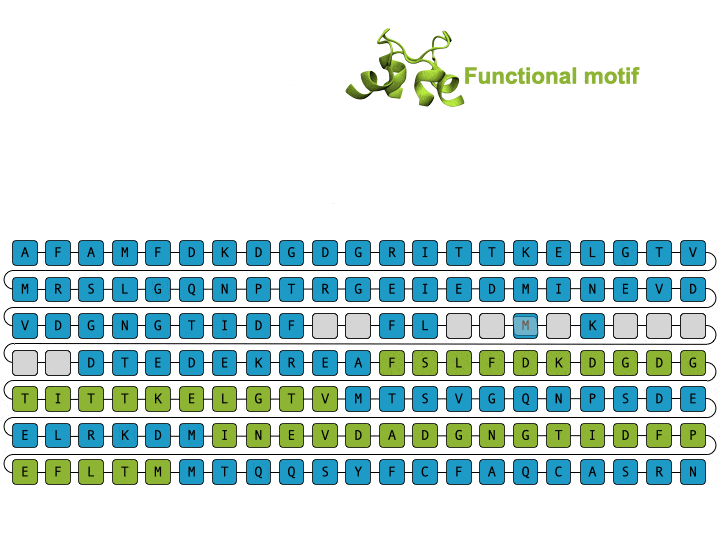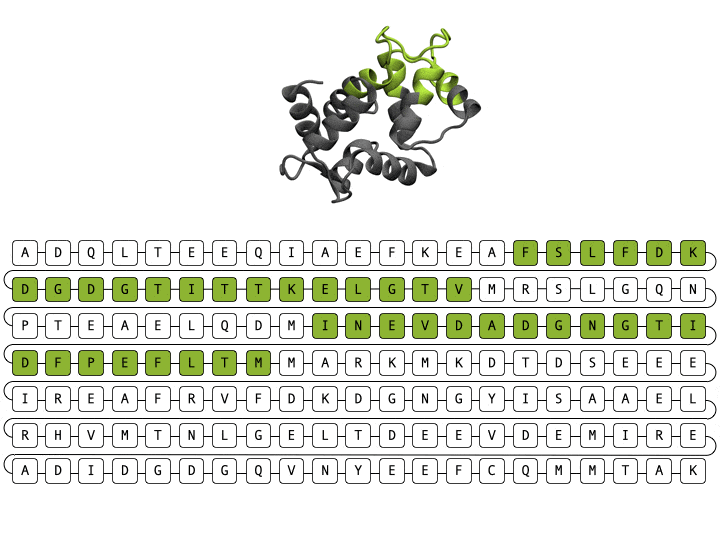
Alamdari et. al., protein sequence and representative structure scaffolding example from deep generative model
Deep generative models are increasingly powerful tools for the in silico design of novel proteins. Recently, a family of generative models called diffusion models has demonstrated the ability to generate biologically plausible proteins that are dissimilar to any actual proteins seen in nature, enabling unprecedented capability and control in de novo protein design. Here, we introduce a general-purpose diffusion framework, EvoDiff, that combines evolutionary-scale data with the distinct conditioning capabilities of diffusion models for controllable protein generation in sequence space. EvoDiff generates high-fidelity, diverse, and structurally-plausible proteins that cover natural sequence and functional space. Critically, EvoDiff can generate proteins inaccessible to structure-based models, such as those with disordered regions, while maintaining the ability to design scaffolds for functional structural motifs, demonstrating the universality of our sequence-based formulation.
Our work featured in TechCrunch:
Microsoft open sources EvoDiff, a novel protein-generating AI | TechCrunch

Interfacial Phenomena
Orientation and Conformation of Proteins at the Air–Water Interface Determined from Integrative Molecular Dynamics Simulations and Sum Frequency Generation Spectroscopy
S. Alamdari*, S. Roeters*, T. Golbek, L. Schmüser, T. Weidner, J. Pfaendtner

Alamdari et al., schematic of SFG+MD evaluation workflow
An air water interface, is one of the simplest representations of an interface in both simulation and the real world. Protein assembly at the air water interface occurs naturally in many biological processes which we know is important to their function. However, the factors that control protein self-assembly at the air water interface, and the dynamic processes that occur during adsorption remain under-explored, and that’s because the introduction of an interface complicates researchers abilities to probe their dynamics and behavior.
In this study, and in this talk, we demonstrate a research approach that combines simulation, theory, and experiment to explore the behavior of large dynamic proteins at an air water interface. We demonstrate that this method allows us to probe protein dynamics and structure with high accuracy, and with this we make some mechanistic interpretations about the behavior of Lysozyme under different pH conditions.
Transcript available on request
Bioinspired Materials
Using simulation tools we are able to explore protein behavior at various interfaces with high specificity and resolution. With this level of resolution, we can link molecular level features (i.e. charge, hydrophobicity, chemistry) to changes in protein structure and orientation at that interface, which we know is highly correlated with protein function.
Non-collagenous bone proteins are important for regulating mineralization of bone in the body. In this work, we use molecular simulations to explore if non-specific adsorption of bone proteins onto implant interfaces provides a plausible mechanism of implant fouling by comparing protein behavior on a model bone surface to a model implant surface. We find that differences in the dominating driving forces at the protein/surface interface lead to differences in their adsorption mechanisms. Knowledge about the forces that drive these assemblies on mineral surfaces, and the mechanisms that change these assemblies on titania can be used to augment the design of new implant coatings.
Impact of glutamate carboxylation in the adsorption of the α-1 domain of osteocalcin to hydroxyapatite and titania
S. Alamdari, J. Pfaendtner

Our work in this area was featured in a UW IT Press Release in 2018